Understanding Genome Assembly in COVID-19 Research
Written on
Chapter 1: The Significance of Genome Assembly
The COVID-19 pandemic has posed significant challenges globally, prompting scientists to work tirelessly to uncover the virus's origin. Recently, researchers unveiled the complete genome of the novel coronavirus. This article explains the process behind this crucial scientific achievement.
What is a Genome?
A genome encompasses all the genetic material of an organism, including every gene. It contains essential information for constructing and sustaining life.
How Sequencing Works
To decode the information embedded in a genome, sequencing is employed. If you’re familiar with DNA analysis from prior articles, you know that sequencing allows us to determine the order of genes, chromosomes, or entire genomes.

Fig 1. A PacBio sequencing machine, a third-generation technology that generates long reads. Image by KENNETH RODRIGUES from Pixabay (CC0).
Sequencing machines extract short random sequences from the genome of interest. Current technologies cannot sequence the entire genome simultaneously; they read small segments averaging between 50-300 bases (next-generation sequencing) or 10,000-20,000 bases (third-generation sequencing), which are referred to as reads.
For more detailed insights on sequencing viral genomes from clinical samples, consider the following articles:
- A complete protocol for whole-genome sequencing of virus from clinical samples: Application to coronavirus OC43
- Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples
Genome Assembly Process
Once we obtain short segments of the genome, the next step is to combine (or assemble) them based on overlapping sequences to reconstruct the complete genome. This assembly process resembles piecing together a jigsaw puzzle. Specialized software, known as assemblers, organizes these reads by identifying overlaps to create continuous sequences called contigs, which may represent the entire genome or sections of it.
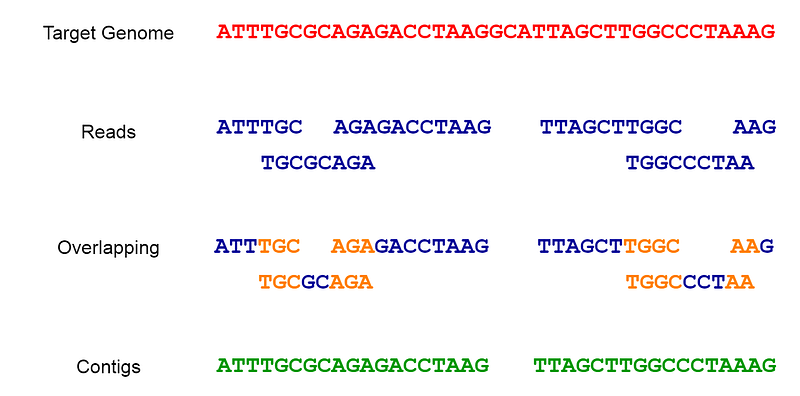
Fig 2. The sequencing and assembly process.
Assemblers are categorized into two main types:
- De novo assemblers: These function without a reference genome (e.g., SPAdes, SGA, MEGAHIT, Velvet, Canu, Flye).
- Reference-guided assemblers: These assemble genomes by mapping sequences to known reference genomes.
Two Primary Assembly Techniques
In bioinformatics, two predominant assembly techniques are recognized:
- Overlap-Layout-Consensus (OLC): This method begins by identifying overlaps among reads, arranging them in a graphical format, and then determining the consensus sequence. SGA is a widely used tool in this category.
- De Bruijn Graph (DBG): Instead of using full reads, this technique segments reads into smaller fragments known as k-mers and constructs a de Bruijn graph from these k-mers. The genomic sequences are then inferred from this graph. SPAdes is a prominent assembler based on the DBG method.
Challenges in Genome Assembly
Genomes possess repetitive patterns of nucleic acids that can complicate the assembly process, leading to ambiguities. Additionally, sequencing machines might not capture every region of the genome, resulting in gaps that can affect the final assembly.
To mitigate these issues, genome assemblers must implement strategies to minimize errors during the assembly process.
Evaluating the Assembly Quality
Assessing the quality of assemblies is crucial to ensure they meet scientific standards. One popular tool for assembly evaluation is QUAST, which utilizes several criteria:
- N50: The minimum contig length necessary to cover 50% of the total assembly.
- L50: The number of contigs longer than N50.
- NG50: Minimum contig length needed to cover 50% of the reference genome.
- LG50: The number of contigs longer than NG50.
- NA50: Minimum length of aligned blocks needed to cover 50% of the total assembly.
- LA50: The number of contigs longer than NA50.
- Genome fraction (%): Percentage of bases aligning to the reference genome.
Getting Started with Genome Assembly
Now, let’s dive into practical experiments using the SPAdes assembler to process reads from patient samples. SPAdes is compatible with next-generation sequencing data. You can also download the QUAST tool for assembly evaluation.
To check if the tools are functioning correctly, run the following commands:
<your_path_to>/SPAdes-3.13.1/bin/spades.py -h
<your_path_to>/quast-5.0.2/quast.py -h
Data Acquisition
You should be familiar with downloading data from the National Center for Biotechnology Information (NCBI). If you need guidance, refer to this link.
The reads for our analysis can be retrieved from NCBI using accession number SRX7636886. Specifically, download the run SRR10971381, which contains data from an Illumina MiniSeq run in FASTQ format. The file will be named sra_data.fastq.gz. Use gunzip to extract the FASTQ file.
Once extracted, you can count the number of reads in your dataset with the following command:
grep '^@' sra_data.fastq | wc -l
This will reveal a total of 56,565,928 reads.
Additionally, download the complete COVID-19 genome from NCBI using GenBank accession number MN908947, which will be in FASTA format and renamed to MN908947.fasta.
Assembling the Reads
To assemble the COVID-19 reads, execute this command in SPAdes:
<your_path_to>/SPAdes-3.13.1/bin/spades.py --12 sra_data.fastq.gz -o Output -t 8
While this demonstration utilizes the general SPAdes assembler, since our reads are RNA-Seq data, it’s advisable to employ the --rna option in SPAdes.
In the Output directory, you’ll find a file named contigs.fasta, which contains the final assembled contigs.
Evaluating the Results
Run QUAST on the assembled contigs with the following command:
<your_path_to>/quast-5.0.2/quast.py Output/contigs.fasta -l SPAdes_assembly -r MN908947.fasta -o quastResult
Viewing the Evaluation Report
After QUAST completes its analysis, navigate to the quastResult folder to review the evaluation outcomes. Open the report.html file in your web browser to see a report similar to that shown in Fig 3. For further information, you can access the "Extended report" section for metrics like NG50 and LG50.
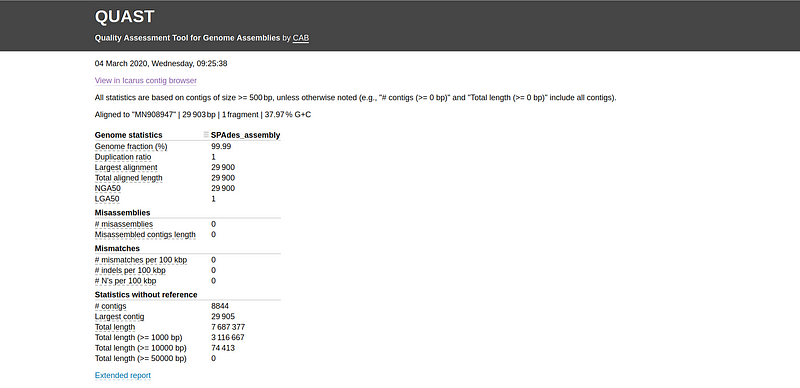
Fig 3. Sample report generated by QUAST.
Analyze the various evaluation metrics, including genome fraction, NG50, NA50, misassemblies, and the number of contigs. You can also explore the alignment of contigs to the reference genome using the Icarus contig browser (select "View in Icarus contig browser") as depicted in Fig 4.
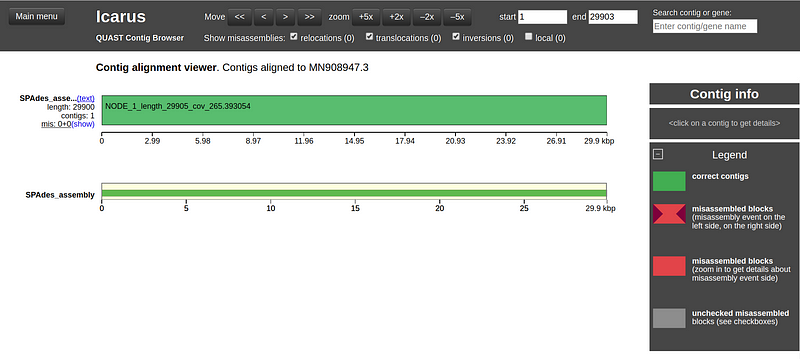
Fig 4. Analysis of contig alignment using the Icarus contig browser.
The Icarus contig browser reveals that the contig labeled NODE_1 aligns closely with the COVID-19 reference genome, achieving a genome fraction of 99.99%. The total aligned length of 29,900 base pairs is nearly identical to the reference length of 29,903 base pairs.
Visualizing the Assembly Graph
For visualizing the assembly graph, the Bandage tool can be employed. Download the precompiled binaries from their official site and open the graph file assembly_graph_with_scaffolds.gfa, which resides in the SPAdes output folder. Load it into Bandage and click on "Draw graph" to visualize the assembly as shown in Fig 5. The prominent green curve in the first row of segments corresponds to NODE_1 from our SPAdes assembly.

Fig 5. Visualization of the SPAdes assembly graph for the COVID-19 dataset.
How Scientists Initially Determined the COVID-19 Genome
In the absence of an exact reference genome for COVID-19, researchers employed metagenomic techniques to analyze viral genomes. They assessed the coverage of contigs (average number of reads covering each base in a contig) and compared them to a bat SARS-like coronavirus isolate, bat SL-CoVZC45 (GenBank accession number MG772933). Their findings indicated that the longest assembled contig exhibited high coverage, closely related to bat SL-CoVZC45, leading to further confirmation tests.
Final Thoughts
Genome assembly has revolutionized our understanding of the genetic makeup of organisms. During the COVID-19 outbreak, it was instrumental in identifying the genetic code of the virus.
The COVID-19 genome comprises 29,903 base pairs (approximately 30k base pairs). With advancements in third-generation sequencing technologies, we may soon be able to sequence entire small genomes directly, reducing the need for assembly. As read lengths increase, direct genome acquisition from sequencing machines will become feasible, especially in metagenomics, where genome sizes vary from kilobases to megabases. Moreover, innovative nanotechnology-based methods, such as quantum sequencing technologies and graphene nanodevices, may soon become prevalent.
Thank you for reading! If you found this article insightful, please share it with your networks. I welcome your thoughts and feedback.
Cheers!
References
Fundamentals of Genome Assembly - In this video, learn the essential concepts behind genome assembly and its significance in genomic studies.
The Fundamentals of Genome Assembly - This video provides a detailed overview of genome assembly techniques and their applications in research.